Durchbruch in der Leukämie-Forschung
Max-Planck-Forscher entdecken, dass die Akute Lymphatische Leukämie (ALL) bei Kindern durch das Fehlen eines speziellen Proteins infolge fehlerhafter Genablesung verursacht wird
Leukämien sind bösartige Erkrankungen des Knochenmarks, bei denen unreife Vorläuferzellen der weißen Blutkörperchen entarten und sich dann unkontrolliert vermehren. Bei etwa 80 Prozent der Leukämien im Kindes- und Jugendalter handelt es sich um die Akute Lymphatische Leukämie (ALL). Dabei verdrängt die ungehemmte Teilung der funktionsuntüchtigen Vorläuferzellen die normalen blutbildenden Zellen und die Entstehung normal funktionierender roter und weißer Blutkörperchen und Blutplättchen wird mehr und mehr vermindert. Bisher war nicht bekannt, wie es zu diesem Verlust der Wachstumskontrolle in den Vorläuferzellen kommt. Wissenschaftler des Max-Planck-Instituts für Immunbiologie und der Universität Freiburg haben jetzt entdeckt, dass bei etwa 50 Prozent dieser Tumor-Erkrankungen ein bestimmtes Protein, das Adapterprotein SLP-65, nicht mehr hergestellt wird. Am Modell der Maus sowie an menschlichen Zellen konnten sie nachweisen, dass der Verlust des SLP-65 zu einem verstärkten Wachstum der weißen Blutzellen und zur Tumorbildung führt. Diese Ergebnisse wurden am 22. Mai 2003 in der Fachzeitschrift "Nature" veröffentlicht.
B-Zellen sind ein wichtiger Teil unseres Immunsystems - sie produzieren Antikörper, die uns vor Infektionen schützen. B-Zellen werden in unserem Körper ständig und in großen Mengen aus so genannten Stammzellen und Vorläufer B-Zellen (prä-B-Zellen) gebildet. Die dafür typische häufige Zellteilung macht diese Zellen besonders anfällig für Entartungen, die zu Tumoren (Leukämien) führen können. Die prä-B-Zell Akute Lymphatische Leukämie (prä-B-ALL) ist die am häufigsten vorkommende Leukämie bei Kindern. Bisher hatte man angenommen, dass diese Leukämie durch Veränderungen im Genom der Vorläuferzellen entsteht. Doch diese Veränderungen sind nicht in allen Fällen nachweisbar und könnten auch erst nach der Entartung der Vorläuferzellen entstanden sein.
Ein Forscherteam um Dr. Hassan Jumaa und Dr. Michael Reth vom Institut für Biologie III der Universität Freiburg und dem Max-Planck-Institut für Immunbiologie in Freiburg haben jetzt herausgefunden, dass in bis zu 50 Prozent der prä-B-ALL Tumore das Adapterprotein SLP-65 fehlt. Dieses Protein kommt normalerweise in allen B-Zellen vor und spielt eine wichtige Rolle bei der Signalleitung vom B-Zell-Antigenrezeptor. Adapterproteine sind Gerüstproteine, die mehrere intrazelluläre Signalproteine zu einem größeren Proteinkomplex zusammenfassen, um die schnelle Verarbeitung intrazellulärer Signale zu ermöglichen. Der vom Adapterprotein SLP-65 aufgebaute Proteinkomplex reguliert die weitere Entwicklung der B-Zellen. Die Forscher stellten nun fest, dass bei Mäusen, denen das SLP-65 Gen fehlt, die Entwicklung der B-Zellen blockiert wird. Doch überraschend fanden die Forscher bei einem Teil dieser SLP-65-defizienten Mäuse nicht nur einen Entwicklungsstopp der prä-B-Zellen, sondern auch leukämieartige prä-B-Zell-Tumore. Die Forscher führten daraufhin das Gen für dieses Protein wieder in die entarteten B-Zellen ein und konnten so das Entstehen der prä-B-Zell-Leukämie in diesen Mäusen verhindern. SLP-65 fördert also nicht nur die B-Zellreifung, sondern verhindert auch ein unkontrolliertes Wachstum dieser Vorläuferzellen. Von daher handelt es sich um ein bisher noch nicht bekanntes Tumorsuppressor-Gen.

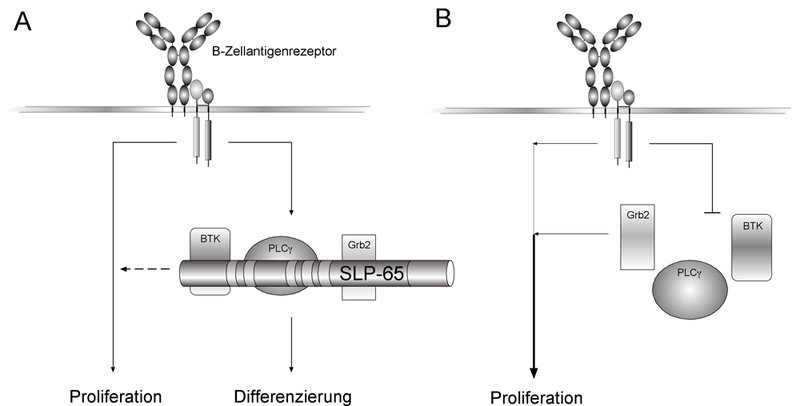
Nach dieser überraschenden Entdeckung fragten sich die Freiburger Max-Planck-Forscher, ob das SLP-65-Gen möglicherweise auch eine Rolle bei menschlichen B-Zell-Leukämien spielen könnte. Sie untersuchten deshalb in Zusammenarbeit mit Kinderkliniken in Freiburg (Dr. C. Niemeyer), Hannover (Dr. M. Schrappe), Zürich, Hamburg und Kuopio (Dr. Pelkonen, Finnland) eine Vielzahl von Leukämie-Proben des Typs prä-B-ALL von zumeist jungen Patienten. Sie stellten dabei fest, dass tatsächlich in fast der Hälfte dieser prä-B-ALL-Tumore das Adaptorprotein SLP-65 fehlte.
Doch wie kann es dazu kommen, dass diese Vorläuferzellen das Adapterprotein nicht mehr produzieren? Auch hier fanden die Forscher die Ursache: Die meisten Gene des Menschen sind in Genfragmente, so genannte Exone, unterteilt. Bevor von diesem Gen ein Produkt, also ein Protein, hergestellt werden kann, muss die Geninformation erst einmal in eine Ribonukleotidsäure (RNS) umgeschrieben werden. Dazu werden zuvor die einzelnen Exone dieses Gens in einem Prozess, den man "Splicing" nennt, zu einer kompletten Boten-RNS (mRNS) zusammengefügt. Das Gen für das Adapterprotein SLP-65 besteht aus insgesamt 17 Exonen, die während der RNS-Prozessierung zu einem Leseraster zusammengefasst werden müssen.

