Mechanism of alternative RNA processing in neurons
Hilgers Lab
One aim of our lab is to obtain mechanistic insight into the co-transcriptional processes that give rise to neuron-specific RNA sequences in neurons.
Most metazoan genes express multiple transcript isoforms through the use of alternative polyadenylation (poly(A)) sites that signal transcription termination. In animals from flies to humans, hundreds of genes undergo a shift toward the distal poly(A) site exclusively in neurons, giving rise to sometimes extremely long 3’UTRs.
We found that the highly conserved neuronal RNA-binding protein ELAV globally regulates all sites of neuronal 3’end processing and directly binds to proximal polyadenylation sites of target mRNAs in vivo. ELAV also regulates neuron-specific alternative splicing, and effectively represents a central effector of neuron- specific transcriptome signatures.
, ELAV binds to the fne RNA and inhibits the mini-exon that encodes a signal for nuclear localization. In the absence of ELAV (Δelav mutant), FNE is activated to replace ELAV function. When both proteins are missing (ΔelavΔfne mutant), neurons lose all their unique 3’UTRs.")
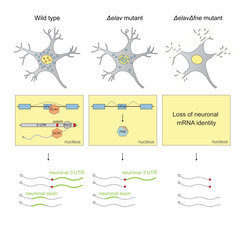
Schematic of the EXAR mechanism in neurons.
Under normal conditions (wild type), ELAV binds to the fne RNA and inhibits the mini-exon that encodes a signal for nuclear localization. In the absence of ELAV (Δelav mutant), FNE is activated to replace ELAV function. When both proteins are missing (ΔelavΔfne mutant), neurons lose all their unique 3’UTRs.
This monopoly of ELAV function can be rescued, if ELAV is impaired, by its paralogue FNE: in elav mutants, the normally cytoplasmic protein FNE enters the nucleus and takes over the functions of ELAV in a novel process termed “Exon-Activated functional Rescue” (EXAR). Neurons lacking both ELAV and FNE proteins, completely lose their neuronal RNA signatures.
We are now focusing on gene-regulatory mechanisms that lead to the emergence of diverse neuron-specific RNA sequences, including ultra-long 3’UTRs, neuron-specific exons, and circular RNAs.

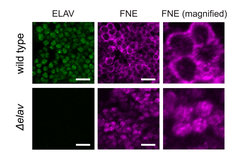
Activated FNE enters the nucleus only in the absence of ELAV. Microscopy image of neuronal cells in Drosophila embryos. ELAV and FNE proteins are made visible by immunohistochemistry. Scale bars: 10µm
Our previous work uncovered an unexpected link between transcription initiation and alternative mRNA processing: ELAV binds to promoter regions of its target genes, and this association is facilitated by promoter-proximal RNA Polymerase II (Pol II) pausing. How ELAV at transcription initiation affects RNA processing many kilobases downstream remains mysterious.
We hypothesize that epigenetic marks, promoter sequence, and Pol II pausing cooperate with ELAV activity to promote the expression of neuron-specific RNAs. To study the regulatory links between transcriptional, co-transcriptional and post-transcriptional processes, we employ functional genetics, RNA biochemistry and proteomics in fly tissues, as well as whole-genome transcriptomics including ChIP, CLIP, and ultra-long-read sequencing.