
Sample preparation
Deep Sequencing Facility
Prior to sequence analysis, any sample must be prepared. It is crucial is to preserve biological information during the complete preparation workflow.
The Deep Sequencing Facility supports sample preparation for multiple assays. Most common types are transcriptome, chromatin, whole genome, and methylome assays. Input material and requirements vary per assay. For each assay, standard, low, or even single-cell preparation technologies are available.
Furthermore, preparation assays differ between short- and long-read sequencing technologies. For short-read sequencing (Illumina), nucleic acids are during preparation artificially fragmented. Long-read sequencing protocols aim to preserve the length of the nucleic acids.
The following section gives an overview of the main preparation techniques in use. Please be aware that there are a vast number of different preparation workflows. Still, most of the published techniques are, to some extend, modifications of the main technologies used in the field. Especially downstream of every workflow (sequencing library preparation), concepts and techniques in use can be quite redundant.
Preparation workflows depend on the RNA fraction of interest.
For genexpression analysis, poly-A containing messenger RNA (mRNA) molecules are purified by using poly-T oligos bound to magnetic beads. To analyze coding and non-coding RNA molecules, ribosomal RNA (rRNA) is depleted using target-specific oligos.
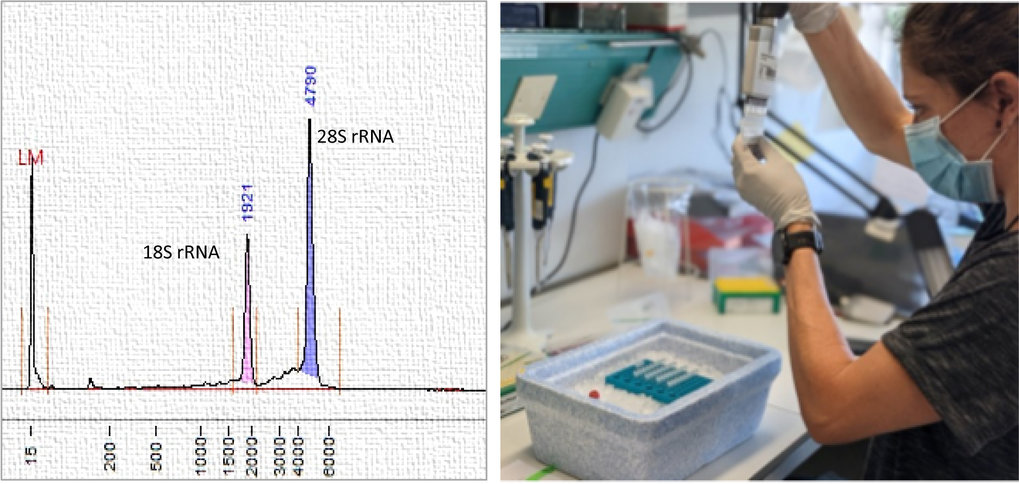
Left: Analysis of high-quality RNA. Right: Preparation of RNA samples for sequencing.
After a capture of polyadenylated mRNAs or a depletion of rRNA background, full-length double-stranded cDNA is prepared. For short-read sequencing (Illumina), cDNA is fragmented, adapter-ligated, and PCR-amplified. For long-read sequencing (Oxford Nanopore), intact cDNA is adapter-ligated and (if needed) PCR-amplified. For both sequencing technologies (long and short-read), multiple indexing options are available for optimizing the use of sequencing capacities and refining sequence accuracy (usage of unique molecular identifiers, UMIs).
For small-RNA sequencing (micro RNAs (miRNAs), small non-coding RNAs), adapters are ligated to each end of the RNA molecule, reverse transcribed, and PCR-amplified. A gel purification step (BluePippin) is necessary to clear libraries from byproducts.
Typically, protein–DNA interactions (ChIP-Seq, CUT&RUN), open chromatin regions (ATAC-Seq), or chromatin conformation (HiC) is analyzed.
For chromatin immunoprecipitation (ChIP-Seq), crosslinking preserves protein–DNA interactions. Upon chromatin isolation, target-specific antibodies are used to select regions of interest. Subsequently, DNA fragments are released and, during library preparation, adapter-ligated and PCR-amplified.
CUT&RUN (Cleavage Under Targets & Release Using Nuclease) works on native chromatin and utilizes target-specific primary antibodies and pAG-MNase (fusion of proteins A and G to micrococcal nuclease) to isolate protein–DNA complexes for analysis using next-generation sequencing.
ATAC-Seq is a technique used to mark transposase-accessible chromatin (profile open chromatin regions). In ATAC-Seq, genomic DNA is exposed to Tn5, a highly active transposase. Tn5 simultaneously fragments DNA and adds sequencing primers (a process known as tagmentation).
HiC is used to study chromosome interactions. Technically: crosslinking of chromatin is followed by restriction enzyme digestion and biotinylation of overhangs before the ligation of nearby ends. Biotinylated ligation junctions are purified and DNA is released for library preparation.
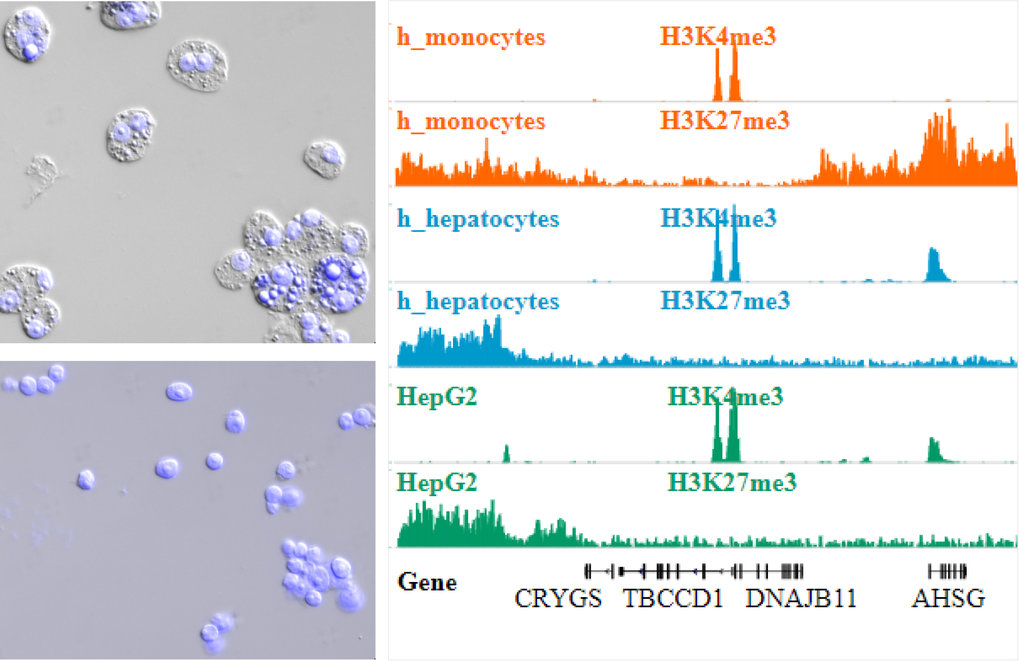
ChIP-Seq including nuclei extraction by sonication (NEXSON) for chromatin preparation. Top left: HepG2 cells before nuclei extraction. Bottom left: HepG2 cells after nuclei extraction. (Pictures in DAPI (blue, nuclei) and differential interference contrast (DIC) channels were taken before and after ultrasound treatment). Right: Read coverage profiles of histone modification ChIP-seq for human monocytes (orange), hepatocytes (blue), and HepG2 cell line (green) prepared by NEXSON.
WGS is a comprehensive method to analyze the entire genome. The technique is rather simple. High-molecular-weight genomic DNA (HMW gDNA) is isolated from material of interest. For short-read sequencing, DNA is fragmented using ultrasound (< 1kb) and sequencing adapters are ligated. Assays in use are sensitive enough to omit PCR amplification prior sequencing.
If Oxford Nanopore Technology (ONT) is used for long-read genome sequencing, read length is limited only by the molecule length in the sample. Therefore, preserving the intactness of DNA prior sequencing adapter-ligation and sequencing is key. Depending on the DNA extraction method, ONT data can be considered “standard long read” (10–100kb) and “ultra-long read” (greater than 100kb).

Left: DNA extraction using Nanobind magnetic discs (circulomics) allows for the extraction of high molecular weight (50–300kb) and ultra high molecular weight (50kb–1Mb) DNA. Right: Loading high molecular weight DNA on a GridION flowcell for long-read sequencing (Oxford Nanopore Technology).
Methyl-Seq is used to detect patterns of DNA methylation. Bisulfite treatment is gold standard for methylome analysis. Treatment with bisulfite converts cytosine residues to uracil but leaves 5-methylcytosine (5-mC) unaffected. Hence, bisulfite treated DNA retains only methylated cytosines.
Using Oxford Nanopore Technology (ONT), epigenetic modifications such as methylation pattern can be directly detected in DNA and RNA molecules, making this technology extremely valuable.
For single cell assays, the 10X Genomics technology is applied. Innovative barcoding concepts facilitate single-cell gene expression, single-cell ATAC, and single-cell multiome (gene expression + ATAC from the same cell) analysis. The 10X technology relies on partitioning thousands of cells into nanoliter-scale gel beads-in-emulsion (GEMs). All generated cDNA molecules share a common 10x barcode. Dual indexed libraries are generated and sequenced and 10x barcodes are used to associate individual reads back to the individual partitions.

10X Genomics technology for single cell analysis. Left: Chromium controller (10X Genomics), Right: GEMs (gel beads-in-emulsion, microscopy picture.)